Болезнь шарко-мари-тута: наследственное заболевание
Содержание:
Как проявляется болезнь Шарко-Мари-Тута
Болезнь Шарко-Мари-Тута проявляется даже в пределах одной семьи не всегда одинаково. И дело не в многообразии её признаков. А в том, что гены, кодирующие данную патологию, способны с разной степенью выраженности формировать симптомы. Проще говоря, имея идентичную «поломку» в хромосомах, признаки недуга у отца и сына будут иметь индивидуальную окраску.
Общие симптомы
Клиническая картина заболевания практически не зависит от его типа и включает:
- атрофию мышц дистальных, то есть наиболее отдалённых от туловища, отделов конечностей;
- снижение сухожильных и периостальных рефлексов;
- изменение чувствительности, характеризующееся её выпадением, но никогда не сопровождающееся появлением ощущения покалывания или «ползания мурашек»;
- деформацию опорно-двигательного аппарата – сколиоз, увеличение свода стопы и др.
Но всё же существует ряд признаков несколько отличающих течение заболевания при различных его вариантах.
Первый тип
Болезнь Шарко-Мари-Тута первого типа нередко протекает в исключительно стёртой форме, при которой пациенты не ощущают изменений в организме и не обращаются за медицинской помощью вообще. Если же патология себя проявила, то происходит это в период первого, максимум второго, десятилетия жизни.
При этом наблюдаются:
- болезненные судороги в мышечном массиве голени, причём редко в икроножной мышце, чаще в передней группе мышц. Подобные спазмы нарастают после периода длительной физической нагрузки (ходьбы, занятий спортом, езде на велосипеде);
- изменения в походке, связанные с постепенным усилением слабости в мышцах. При этом у детей это может дебютировать в хождении на цыпочках;
- деформация стоп с формированием высокого свода последних и наличием молоточкообразных пальцев, которая развивается как результат дисбаланса в тонусе сгибателей и разгибателей;
- атрофия мышц, начинающаяся со стоп и поднимающаяся на голень. Затем процесс затрагивает кисть – появляется тремор в руках и выраженная слабость в пальцах, особенно при попытке выполнять мелкие движения. К примеру, застёгивать пуговицы, перебирать крупу;
- угнетение или полное отсутствие сухожильных и периостальных рефлексов, а именно ахиллового, карпорадиального, при сохранных с более проксимальных отделов рук и ног. То есть коленный и рефлексы с двуглавой и трёхглавой мышцы остаются интактными;
- нарушения чувствительности в кистях и стопах, выражающиеся в её постепенном выпадении. Причём стартует патология с вибрационной и тактильной сферы, распространяясь на суставно-мышечные и болевые ощущения;
- сколиоз и кифосколиоз;
- утолщение нервных стволов, чаще всего поверхностного малоберцового и большого ушного.
Для болезни Шарко характерна атрофия мышц именно в дистальных отделах конечностей. При этом если не выражена подкожно-жировая клетчатка, объём голени и бедра разительно отличается, и ноги приобретают вид таковых у аиста или походят на перевёрнутую бутылку для шампанского.
Невральная амиотрофия Шарко Мари первого типа имеет атипичные формы. Одна из них — синдром Руси-Леви, при котором наблюдается выраженный тремор при попытке удержать руки в одном положении и неустойчивость при ходьбе. Сюда же относится заболевание, проявляющееся, помимо стандартных симптомов, парезами, гипертрофией мышц голени, резким выпадением чувствительности и ночными судорогами в икроножных мышцах.
Второй тип
Для болезни Шарко-Мари-Тута второго типа, кроме более позднего начала, характерны:
- менее выраженные изменения чувствительности;
- более редкая встречаемость деформаций стопы и пальцев;
- наличие синдрома беспокойных ног (возникают неприятные ощущения в ногах во время отхода ко сну, заставляющие пациента двигаться, что облегчает состояние);
- нередко сохранная сила в кисти;
- отсутствие утолщения нервных стволов.
При синдроме Шарко-Мари-Тута, передающемся через Х-хромосому, могут встречаться нейросенсорная тугоухость (снижение слуха) и транзиторная энцефалопатия, возникающая после физической нагрузки на высоте. Для последней характерно появление симптомов через 2-3 дня после занятий. Признаками патологии становятся шаткость, нарушение речи, глотания, слабость в проксимальных отделах рук и ног. Обычно клиническая картина недуга исчезает самостоятельно в течение пары недель.
Лечение болезни Шарко-Мари-Тута
Лечение назначается только после подтверждения диагноза врачом-специалистом. Показаны дозированная ЛФК и массаж, ортопедические мероприятия, витаминные препараты, средства нейротрофического действия, улучшающие микроциркуляцию, антихолинэстеразные препараты.
Основные лекарственные препараты
Имеются противопоказания. Необходима консультация специалиста.
- (средство, улучшающее метаболизм и энергообеспечение тканей). Режим дозирования: в/м, в первые 2-3 дня вводят 1 раз в день по 1 мл 1%-ного раствора, в последующие дни 2 раза в день или сразу 2 мл 1%-ного раствора 1 раз в день. На курс лечения — 30-40 инъекций.
- (средство, улучшающее микроциркуляцию). Режим дозирования: внутрь, проглатывая целиком, во время или сразу после приема пищи, запивая достаточным количеством воды, в дозе 100 мг 3 раза в сутки с последующим медленным повышением дозы до 200 мг 2-3 раза в сутки.
- (комплекс витаминов группы В). Режим дозирования: терапию начинают с 2 мл внутримышечно 1 р/д на протяжении 5-10 дней. Поддерживающая терапия — 2 мл в/м два или три раза в неделю.
- (анаболическое стероидное средство). Режим дозирования: внутрь, перед едой в дозе 0,005-0,01 г 1-2 раза в день. Курс лечения у взрослых длится 4-8 недель. Перерывы между курсами 4-8 недель.
- (ноотропное средство). Режим дозирования: применяют парентерально в виде в/м инъекций (до 5 мл) и в/в инъекций (до 10 мл). Препарат в дозе от 10 мл до 50 мл рекомендуется вводить только посредством медленных в/в инфузий после разведения стандартными растворами для инфузий. Продолжительность инфузий составляет от 15 до 60 мин. Вводят парентерально в дозе от 5 мл до 30 мл/сут. Рекомендуемый оптималь-ный курс лечения — ежедневные инъекции в течение 10-20 дней.
- (антихолинэстеразное средство). Режим дозирования: внутрь, суточная доза для взрослых составляет 10-40 мг в 2-4 приема.
Наследственное заболевание. Основной тип передачи – аутосомно-доминантный (с пенетрантностью патологического гена около 83%), реже – аутосомно-рецессивный.
Морфологическую основу болезни составляют дегенеративные изменения главным образом в периферических нервах и нервных корешках, касающиеся как осевых цилиндров, так и миелиновой оболочки. Иногда наблюдаются гипертрофические явления в интерстициальной ткани. Изменения в мышцах носят преимущественно неврогенный характер, отмечается атрофия отдельных групп мышечных волокон; в неатрофированных мышечных волокнах структурные изменения отсутствуют. По мере прогрессирования заболевания появляются гиперплазия интерстициальной соединительной ткани, изменения в мышечных волокнах – их гиалинизация, центральное смещение ядер сарколеммы, гипертрофия некоторых волокон. В более поздних стадиях болезни отмечаются гиалиновая дегенерация, распад мышечных волокон. Наряду с этим в ряде случаев отмечены изменения в спинном мозге. Они складываются из атрофии клеток передних рогов, главным образом в поясничной и шейной части спинного мозга, и различной степени поражения проводниковых систем, характерного для наследственной атаксии Фридрейха.
Диагностика
Диагностическая процедура включает ряд методов, среди которых:
- подробный личный и семейный анамнез;
- клиническая оценка мышечной силы, чувствительности;
- электрофизиологическое исследование скорости проводимости нервного волокна;
- неврологическое исследование.
Наиболее распространенные формы заболевания могут быть диагностированы путем анализа ДНК из крови пациента.
При диагностике необходимо тесное сотрудничество невролога, генетика, реабилитолога, хирурга-ортопеда и протезиста. В соответствии с выводами обследования, даются рекомендации относительно индивидуального плана реабилитации, при необходимости, назначается ортопедическая операция.
Значительная изменчивость клинических признаков болезни, наряду с отсутствием знаний о ней у многих врачей, часто приводит к неправильной диагностике.
Причины
Болезнь Шарко-Мари-Тута была открыта в далеком 1847 году, а описание клинической картины появилось в 1869 году. Но наиболее тщательно недуг стали изучать в 90-е годы XX столетия, когда синдром стал появляться все чаще.
Как уже говорилось выше, данный вид патологии является наследственным, хотя были зафиксированы случаи спорадического характера, которые установили, что главной причиной возникновения недуга является мутация в генах.
Исходя из имеющихся данных, подобные мутации были зафиксированы у 20% семей с синдромом Шарко. Недуг относится к самым частым патологиям, способных прогрессировать, а также это один из самых тяжелых дегенеративных процессов в ЦНС. Причиной развития синдрома Шарко также называют инфекционно-токсические заболевания, которые вызывает вирус с большой нейротропностью.
Список литературы
- МКБ-10 (Международная классификация болезней)
- Юсуповская больница
- Батуева Е.А., Кайгородова Н.Б., Каракулова Ю.В. Влияние нейротрофической терапии не нейропатическую боль и психовегетативный статус больных диабетической нейропатией // Российский журнал боли. 2011. № 2. С. 46.
- Бойко А.Н., Батышева Т.Т., Костенко Е.В., Пивоварчик Е.М., Ганжула П.А., Исмаилов А.М., Лисинкер Л.Н., Хозова А.А., Отческая О.В., Камчатнов П.Р. Нейродикловит: возможность применения у пациентов с болью в спине // Фарматека. 2010. № 7. С. 63–68.
- Морозова О.Г. Полинейропатии в соматической практике // Внутренняя медицина. 2007. № 4 (4). С. 37–39.
Симптомы
Невральная амиотрофия Шарко-Мари-Тута начинается с развития симметричных мышечных атрофий в дистальных отделах ног. Начальные симптомы манифестируют, как правило, в первой половине второго десятилетия жизни, реже в период от 16 до 30 лет. Они заключаются в повышенной утомляемости стоп при необходимости длительно стоять на одном месте. При этом наблюдается симптом «топтания» – чтобы снять утомляемость стоп пациент прибегает к ходьбе на месте. В отдельных случаях невральная амиотрофия манифестирует расстройствами чувствительности в стопах, наиболее часто — парестезиями в виде ползания мурашек. Типичным ранним признаком ШМТ является отсутствие ахилловых, а позже и коленных сухожильных рефлексов.
Развивающиеся первоначально атрофии затрагивают в первую очередь абдукторы и разгибатели стопы. Результатом является свисание стопы, невозможность ходьбы на пятках и своеобразная походка, напоминающая вышагивание лошади, — степпаж. Далее поражаются приводящие мышцы и сгибатели стопы. Тотальная атрофия мышц стопы приводит к ее деформации с высоким сводом, по типу стопы Фридрейха; формируются молоткообразные пальцы стопы. Постепенно атрофический процесс переходит на более проксимальные отделы ног — голени и нижние части бедер. В результате атрофии мышц голени возникает болтающаяся стопа. Из-за атрофии дистальных отделов ног при сохранности мышечной массы проксимальных отделов ноги приобретают форму перевернутых бутылок.
Зачастую при дальнейшем прогрессировании болезни Шарко-Мари-Тута атрофии появляются в мышцах дистальных отделов рук — вначале в кистях, а затем и в предплечьях. Из-за атрофии гипотенара и тенара кисть становиться похожей на обезьянью лапу. Атрофический процесс никогда не затрагивает мышцы шеи, туловища и плечевого пояса.
Часто невральная амиотрофия Шарко-Мари-Тута сопровождается легкими фасцикулярными подергиваниями мышц рук и ног. Возможна компенсаторная гипертрофия мышц проксимальных отделов конечностей.
Сенсорные нарушения при невральной амиотрофии характеризуются тотальной гипестезией, однако поверхностная чувствительность (температурная и болевая) страдает значительно больше глубокой. В некоторых случаях наблюдается цианоз и отек кожи пораженных конечностей.
Для болезни Шарко-Мари-Тута типично медленное прогрессирование симптомов. Период между клинической манифестацией заболевания с поражения ног и до появления атрофий на руках может составлять до 10 лет. Несмотря на выраженные атрофии, пациенты длительное время сохраняют работоспособное состояние. Ускорить прогрессирование симптомов могут различные экзогенные факторы: перенесенная инфекция (корь, инфекционный мононуклеоз, краснуха, ангина, ОРВИ), переохлаждение, ЧМТ, позвоночно-спинномозговая травма, гиповитаминоз.
Спинальные амиотрофии
Спинальная амиотрофия представляет собой прогрессирующее заболевание в результате которого поражаются нервные клетки спинного мозга. Это не одно заболевание, сюда входит целая группа болезней: болезнь Арана-Дюшена, болезнь Вердинга-Гоффмана и ряд других более редких заболеваний.
Несмотря на многочисленность заболеваний, входящих в данную группу, все они проявляются сходными симптомами. Это выражается в том, что со временем развиваются вялые параличи, ослабевают сухожилия. Как правило, поражения ассиметричны. Особенности каждого заболевания в том, что страдают вначале различные группы мышц.
Например, при заболевании Вердинга-Гоффмана у больного наблюдается слабость, в основном страдают мышцы туловища. Исследователи отмечают высокий процент кровного родства среди родителей заболевших. Данное заболевание подразделяется на виды в зависимости от времени появления и прогрессирования заболевания: врожденная, ранняя детская и поздняя.
Развитие врожденной амиотрофии происходит в первые месяцы жизни малыша. Данное заболевание, как правило, сочетается с другими пороками. При запоздалом лечении высока вероятность летального исхода. Причиной последнего является сердечнососудистая и дыхательная недостаточность, которая развивается из-за слабости дыхательных мышц.
Ранняя детская амиотрофия развивается в возрасте от полугода до одного года. Изначально поражаются мышцы туловища и ног, в дальнейшем происходит нарушение работы всех групп мышц. Обнаружить это заболевание достаточно просто.
Малыш не встает на ножки, не может сидеть и с трудом захватывает игрушки. Характерной чертой также является небольшое подергивание мышц, особенно языка. Если вовремя не приступить к лечению, то развивается полная гипотония мышц и паралич. Ребенок с таким заболеванием не доживает до 15 лет.
Поздняя амиотрофия проявляется в возрасте от двух с половиной до трех с половиной лет. В это время ребенок уже устойчиво стоит на ногах и свободно передвигается в пространстве.
Симптомами этого заболевания является неуверенность при ходьбе и частые падения. Данное заболевание постепенно прогрессирует, поражая все большую группу мышц. В итоге к десяти годам ребенок перестает самостоятельно передвигаться и не может себя обслуживать. Прожить с подобным заболеванием человек может лишь максимум до 30 лет.
Доброкачественная спинальная амиотрофия Кугельберга-Веландер. Отдельное заболевание, которое входит в группу болезней спинальной амиотрофии. Отдельная группа исследователей считает, что данное заболевание является разновидностью болезни Вердинга-Гоффмана.
Данное заболевание медленно прогрессирует, развивается, как правило, в мышцах туловища и постепенно распространяется на конечности. Сопровождается общей слабостью. Наблюдается у детей в возрасте от трех до семнадцати лет. Характерной чертой данной болезни является также избыточная масса тела. Люди с подобным заболеваниям доживают до глубокой старости и сохраняют способность самостоятельно передвигаться.
Болезнь Арана-Дюшера наблюдается у лиц в старческом возрасте. Характеризуется ослаблением мышц конечностей. Само течение болезни медленное. Наблюдается подергивание мышц, а в отдельных случаях и параличи. Смерть при данном заболевании наступает от бронхопневмонии.
Осложнения
Если запустить невральную амиотрофию, то могут быть необратимые последствия. Проявляются они в виде нарушения дыхательной системы. Если патология поражает нервные окончания, которые контролируют диафрагму. Больному скорее всего нужно будет употреблять бронхолитические средства или искусственную вентиляцию легких. Лишний вес или сильное ожирение может привести к тому, что пациенту становится трудно дышать.
Депрессивное состояние может быть из-за стрессовых ситуаций. Чаще всего возникает это из-за прогрессирования патологии. Могут применяться, как специальные лекарственные препараты, так и психологическая помощь. Если заболевание будет в запущенной форме и без лечения, то это приведет к инвалидности.

Пациент может перестать полностью передвигаться и на этой фоне возникнуть глухота. Для того чтобы не было такие серьезных последствий, необходимо своевременно обращаться к врачу. Он сможет назначить комплексную диагностику, чтобы поставить точный диагноз. Только после этого будут применяться методы лечения. Заболевание предоставляет хроническую моторную и сенсорную полинейропатию.
Лечение болезни Шарко-Мари-Тута
(с) The New York Times / Michael Nagle
Пока нет никакого лечения для ШМТ, но можно облегчить симптомы и отсрочить начало инвалидности.
НПВС (нестероидные противовоспалительные препараты), такие как ибупрофен, уменьшают суставные и мышечные боли, а также боли, вызванные поврежденными нервами.
Трициклические антидепрессанты (ТЦА) назначают, если НПВС не эффективны. ТЦА обычно используют для лечения депрессии, но они могут уменьшить болевые симптомы невропатии. Тем не менее, они имеют побочные эффекты.
Физическая терапия поможет укрепить и растянуть мышцы. Упражнения помогут сохранить мышечную силу.
Трудотерапия может помочь пациентам, которые имеют проблемы с движениями пальцев и им трудно осуществлять повседневную деятельность.
Ортопедические устройства могут предотвратить травмы. Обувь с высокими голенищами или специальные ботинки обеспечивают дополнительную поддержку голеностопного сустава, и специальная обувь или стельки для обуви могут улучшить походку.
Операция по удалению ахиллова сухожилия иногда может облегчить боль и сделать ходьбу легче. Хирургия может исправить плоскостопие, облегчить боль в суставах.
Методы лечения
Лечение болезни Шарко направлено на уменьшение проявления симптомов. Специальной терапии, которая бы избавила больного от заболевания навсегда, в настоящее время не существует. Предотвратить болезнь невозможно, поскольку она передается генетически.
 Терапевтические методы включают в себя применение препаратов, улучшающих кровообращение и обменные процессы в тканях, коэнзим Q, витамин Е. Для улучшения нервной проводимости популярное лекарство – Галантамин. При болях в ногах назначаются антидепрессанты, противосудорожные препараты. Опытным путем установлена эффективность больших доз витамина С при 1-м типе болезни. Благодаря использованию витамина С удавалось замедлить разрушение миелиновых оболочек нейронов.
Терапевтические методы включают в себя применение препаратов, улучшающих кровообращение и обменные процессы в тканях, коэнзим Q, витамин Е. Для улучшения нервной проводимости популярное лекарство – Галантамин. При болях в ногах назначаются антидепрессанты, противосудорожные препараты. Опытным путем установлена эффективность больших доз витамина С при 1-м типе болезни. Благодаря использованию витамина С удавалось замедлить разрушение миелиновых оболочек нейронов.
При ухудшении состояния больного (чаще всего из-за включения аутоиммунных симптомов) назначаются иммуноглобулины, кортикостероиды (например, Метилпреднизолон). Может проводиться плазмоферез.
Для лечения данного заболевания широко применяются ЛФК, физиотерапевтические методы: массаж, гидромассаж, грязевые ванны, бальнеотерапия и другие
При нарушении сенсорных функций нужно осторожно применять гальванизацию и электрофорез. Полезны занятия плаванием, спортивной ходьбой, но без слишком больших нагрузок
В случаях, когда пациент теряет способность самостоятельно передвигаться, может проводиться хирургическая операция. Чаще всего это артродез голеностопных суставов – сращивание большеберцовой и таранной костей. Это придает ноге неподвижность в голеностопе, но исключает опрокидывание при ходьбе и дает человеку возможность ходить.
Симптомы болезни Шарко Мари Тута медленно прогрессируют, но заметно это не всегда. Ведь порой это происходит на протяжении десятилетий
Важно, что наличие данного заболевания не влияет на продолжительность жизни человека – только на ее качество и самочувствие больного. При правильно и своевременно проведенных лечебных процедурах развитие болезни можно замедлить
Широко применяются в лечении больных различные ортопедические приспособления. Для большей устойчивости голеностопа показано ношение высокой обуви, специальных повязок, которые предупреждают вывихи и растяжения – типичные осложнения этого заболевания.Однако нужно помнить, что запущенная стадия болезни Шарко приводит к инвалидности. При дистрофических процессах в руках больной утрачивает способность выполнять домашние дела, работу руками, не может себя обслуживать. Если страдают мышцы и суставы ног, для передвижения человеку нужны инвалидная коляска, поддерживающие устройства и опоры.
Тем не менее, большинство заболевших адаптируется к жизни, могут себя обслуживать, передвигаться самостоятельно, даже вести трудовую деятельность. Учитывая возможные проблемы в будущем с движениями, координацией, мелкой моторикой, иногда со слухом, надо постараться заранее ориентироваться (либо ориентировать пациента-ребенка) на выбор подходящей профессии.
Процедуры и операции
Особое внимание уделяется немедикаментозным методам терапии. Комплекс мероприятий, позволяющих достичь максимального терапевтического эффекта:
- Лечебная физическая культура. Регулярные занятия ЛФК повышают мышечный тонус. Наибольший эффект достигается при достижении пассивных упражнений (занятия вместе со специалистом) и активных (выполняются самостоятельно).
- Электростимуляция. Направленная подача электрических импульсов улучшает проводимость по периферической нервной системе, активирует метаболические процессы в паретичных мышцах, усиливает нейротрофику.
- Бальнеотерапия. Грязевые аппликации и грязевые ванны замедляют формирование контрактур и улучшают работу вегетативной нервной системы.
- Массаж. Различные виды массажа (аппаратный и ручной) улучшают лимфатический отток и нормализуют кровообращение. Рекомендован расслабляющий, стимулирующий и вибрационный массаж.
- Ортопедическая терапия. Ношение специальной ортопедической обуви позволяет предотвратить развитие грубой деформации. Из-за мышечной слабости развивается нестабильность суставов. Подтяжки, ортезы и другие специальные приспособления используются для фиксации стоп в необходимом положении.
При комплексном подходе к терапии повышается уровень мышечной силы, нормализуется походка и исправляются различные нарушения равновесия. Грамотно подобранное лечение повышает социальную и бытовую адаптацию, восстанавливает работоспособность пациента.
1.2.2. Невральная амиотрофия шарко-мари-тута.
Частота 1:500000 населения. Наследуется по аутосомно-доминантному, аутосомно-рецессивному сцепленному с Х-хромосомой типу. Обнаруживается сегментарная демиелинизация в нервах, в мышцах — денервация с явлениями «пучковой» атрофии мышечных волокон.
КЛИНИКА. Первые признаки заболевания чаще проявляются в 15 — 30 лет, реже в дошкольном возрасте. Характерными симптомами являются мышечная слабость, патологическая утомляемость в дистальных отделах ног. Больные быстро устают при длительном стоянии на одном месте и нередко для уменьшения утомления в мышцах прибегают к ходьбе на месте («симптом топтания«). Реже заболевание начинается с чувствительных расстройств — болей, парестезий, ощущения ползания мурашек. Атрофии первоначально развиваются в мышцах голеней и стоп. Мышечные атрофии, как правило, симметричные. Поражается перонеальная группа мышц и передняя большеберцовая мышца. Вследствие атрофий ноги резко сужаются в дистальных отделах и приобретают форму «перевернутых бутылок» или «ног аиста». Стопы деформируются, становятся «выеденными» с высоким сводом. Парез стоп изменяет походку больных. Они ходят, высоко поднимая ноги; ходьба на пятках невозможна. Атрофии в дистальных отделах рук — мышцах тенара, гипотенара, а также в мелких мышцах кистей присоединяются позже. Ахилловы рефлексы снижаются в ранних стадиях болезни, а коленный рефлекс, рефлекс с трех-двуглавой мышц плеча длительное время остаются сохранными. Чувствительные расстройства объективно определяются нарушениями поверхностной чувствительности по периферическому типу (тип «перчаток» и «носков»). Часто имеются вегетативно-трофические нарушения — гипергидроз стоп и кистей, гиперемия кистей и стоп. Интеллект обычно сохранен.
Течение медленно прогрессирующее. Прогноз благоприятен.
Диагностика. Тип наследования, атрофии дистальных отделов конечностей, расстройства чувствительности по полиневритическому типу, медленное прогрессирующее течение, результаты электромиографии (снижение скорости проведения по периферическим нервам), биопсия нервов.
ЛЕЧЕНИЕ ПМД. Применяют витамины группы В, С Е, а также АТФ, церебролизин, ноотропил, энцефабол, фосфаден, карнитина хлорид, метионин, лецитин, глутаминовая кислота, ретаболил. Положительный эффект дают антихолинэстеразные препараты (прозерин, местинон, галантамин). Показаны средства, улучшающие микроциркуляцию: никотиновая кислота, трентал, пармидин. Наряду с медикаментозной терапией применяют ЛФК, массаж, электрофорез лекарственных средств (прозерин, кальция хлорид), диадинамические токи, синусоидальные модулированные токи, электростимуляция, ультразвук, озокерит, грязевые аппликации, радоновые, хвойные, сульфидные и сероводородные ванны, оксигенобаротерапия. Показано ортопедическое лечение при контрактурах конечностей, умеренной деформации позвоночника и асимметричном укорочении конечностей.
13.2. Семейная атаксия фридрейха.
Наследственное дегенеративное заболевание нервной системы, характеризующееся синдромом поражения задних и боковых столбов спинного мозга. Тип наследования аутосомно-рецессивный с неполной пенетрантностью патологического гена. Мужчины и женщины болеют одинаково часто.
КЛИНИКА. Начало заболевания относится к 6 — 15-летнему возрасту. Первым симптомом болезни является неустойчивая походка. В ранних стадиях атаксия выражена преимущественно в ногах. По мере прогрессирования заболевания нарушения координации распространяются на руки и лицо. При неврологическом обследовании выявляются крупноразмашистый нистагм, атаксия в руках и ногах, адиадохокинез, дисметрия, скандированная речь, расстройства мышечно-суставного чувства и вибрационной чувствительности. Нарушается почерк. Ранним симптомом является снижение, а затем угасание сухожильных и периостальных рефлексов. Мышечный тонус понижен. В более поздних стадиях болезни присоединяются афферентный парез нижних, а затем верхних конечностей, нередко патологические пирамидные рефлексы, дистальные мышечные атрофии. Интеллект снижен. Заболевание медленно прогрессирует. Средняя продолжительность жизни 10 — 15 лет с момента его развития.
Диагностика. Заболевание распознается на основании характерных симптомов — деформаций стоп по типу стопы Фридрейха (высокий свод, экстензия основных фаланг пальцев стопы, флексия концевых фаланг), позвоночника, поражения миокарда, эндокринных расстройств.
ЛЕЧЕНИЕ. Применяются симптоматические средства: общеукрепляющие препараты, ЛФК, массаж. В некоторых случаях производится хирургическая коррекция деформации стоп.
Что это за болезнь
Данный недуг является врожденной патологией строения нервных стволов, что влечет за собой потерю возможности передачи нервных импульсов. Синдром Шарко приводит и к другим неприятным последствиям, например, к потере двигательной функции. Если вовремя не начать терапию заболевания, то синдром начнет прогрессировать. Это чревато хромотой, а в самых тяжких случаях появляется нарушение дыхательной функции.
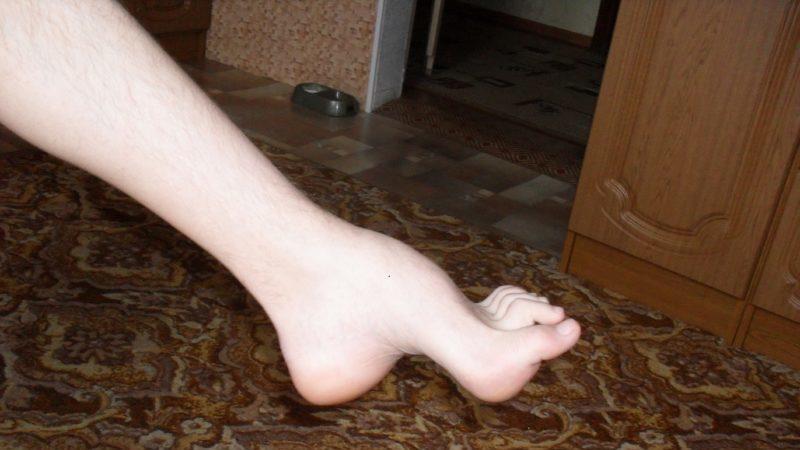
Вышеуказанный недуг имеет и иные названия: патология Тута или Мари. В совокупности эти три врача Шарко-Мари-Тута впервые начали изучение болезни, за что синдром и получил такое название.
Специалистам удалось доказать, что недуг является наследственным. Если хоть один из членов семьи болеет данным синдромом, то вероятность более 50%, что у его детей будет тот же диагноз. Симптомы же могут быть иные.